DAG-Based Batch Optimization for Cohort SQL Generation
Source:vignettes/a07_batch-optimization.Rmd
a07_batch-optimization.RmdOverview
When generating SQL for multiple ATLAS cohort definitions, the usual approach treats each cohort independently – parsing its JSON, expanding its concept sets against the vocabulary, scanning CDM domain tables, building qualified events, and writing results. This works, but it repeats expensive database operations (vocabulary joins, full-table scans, result-table I/O) once per cohort.
atlasCohortGenerator implements a DAG-based
batch optimizer that reorganizes a set of cohort SQL scripts
into a single execution plan with shared intermediate stages. The
optimizer produces semantically identical results to
the one-at-a-time approach while eliminating redundant work.
This vignette explains:
- How the one-at-a-time (CIRCE-based) approach works
- How the optimized batch DAG is structured
- How we guarantee equivalence between the two approaches
- Performance characteristics under different workload types
The One-at-a-Time Approach
The traditional CIRCE approach generates a fully self-contained SQL script for each cohort. Every script independently performs five phases:
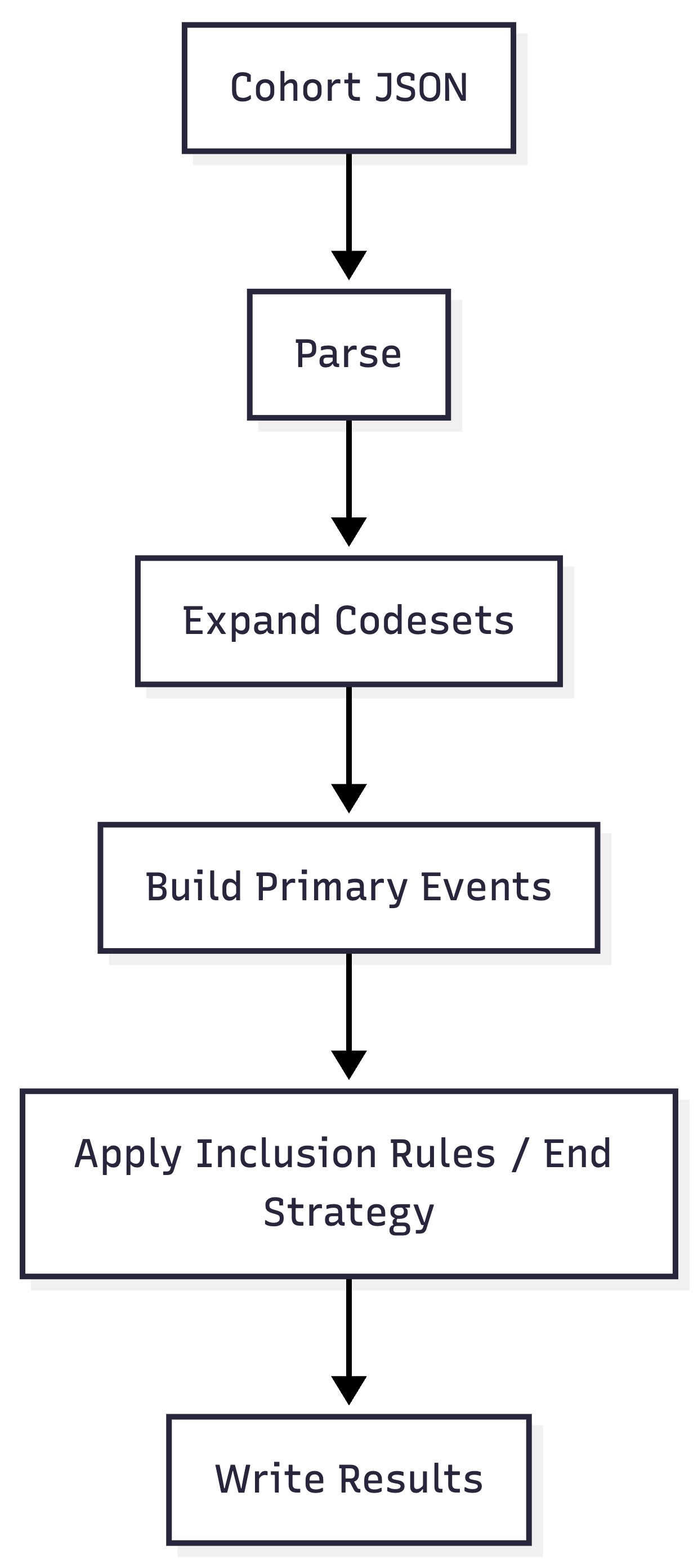
For a batch of N cohorts, the database executes N independent pipelines.
Each cohort creates its own local #Codesets temp table,
joins against CONCEPT_ANCESTOR and CONCEPT to
expand descendants, scans CDM domain tables
(e.g. DRUG_EXPOSURE, CONDITION_OCCURRENCE)
with joins to its local codesets, and writes directly to the results
tables. For N cohorts, the database performs:
- N vocabulary expansions (CONCEPT_ANCESTOR joins)
- N scans per CDM domain table used
- N DELETE + INSERT cycles on each result table
The Optimized Batch DAG
The optimizer restructures these N independent pipelines into a single DAG with shared intermediate stages. The key insight is that vocabulary expansion and CDM table scanning are cohort-independent – we can do them once and share the results.
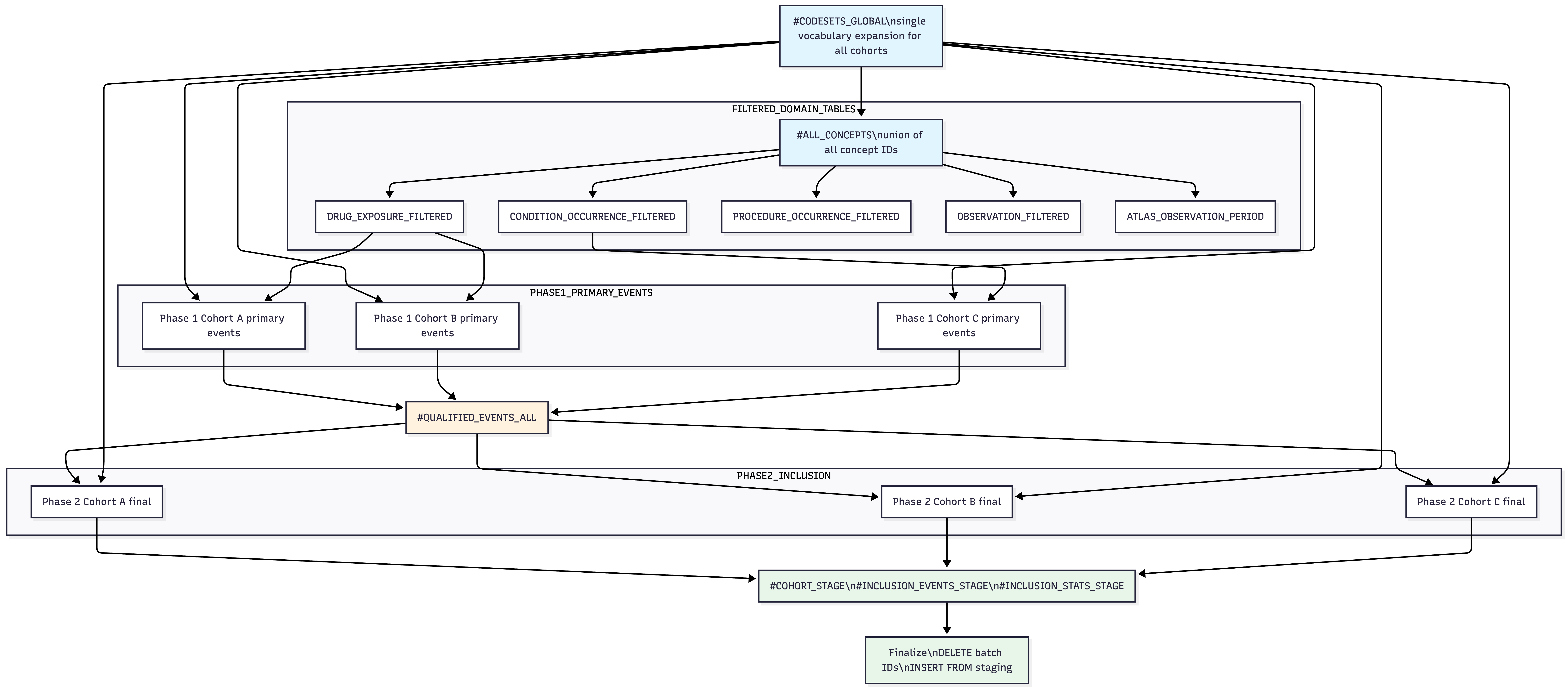
The batch script executes in a strict topological order with no back-edges or cross-cohort dependencies:
| Phase | What happens | Shared? |
|---|---|---|
| 1. Staging setup | Create #COHORT_STAGE,
#INCLUSION_EVENTS_STAGE,
#INCLUSION_STATS_STAGE
|
Once |
| 2. Global codesets | Build #CODESETS_GLOBAL with all cohorts’ concept sets;
extract #ALL_CONCEPTS
|
Once |
| 3. Domain filtering | Create ##DRUG_EXPOSURE_FILTERED,
##CONDITION_OCCURRENCE_FILTERED, etc. |
Once per domain |
| 4. Phase 1 | For each cohort: slice local #Codesets from global,
build primary events into #QUALIFIED_EVENTS_ALL
|
Per-cohort, shared output |
| 5. Phase 2 | For each cohort: slice #qualified_events from
#QUALIFIED_EVENTS_ALL, run inclusion rules, end strategy,
write to staging |
Per-cohort |
| 6. Finalize | Single DELETE by batch IDs + INSERT from staging into result tables | Once |
Worked Example: Three Drug Exposure Cohorts
Consider three cohorts that identify users of different drugs:
- Cohort A: Aspirin users (concept set with aspirin concepts)
- Cohort B: Metformin users (concept set with metformin concepts)
- Cohort C: Aspirin users with a diabetes diagnosis (same primary criteria as A, plus an inclusion rule requiring a condition occurrence)
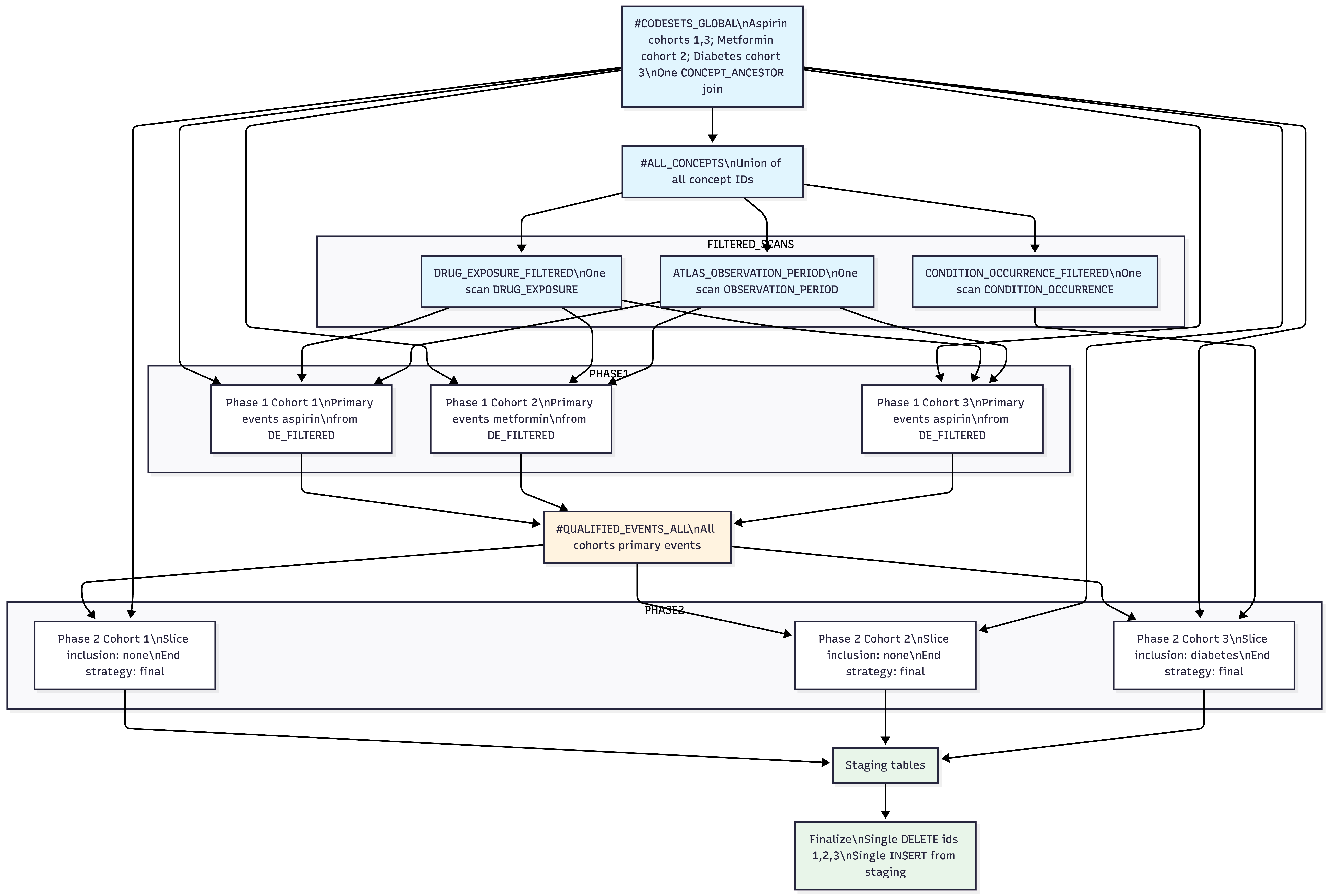
How Equivalence Is Guaranteed
The batch optimizer must produce exactly the same cohort membership as running each cohort independently. We guarantee this through four mechanisms:
1. Same Builder, Different Assembly
Both paths use the same
build_cohort_query_internal() function to generate
per-cohort SQL. The batch path does not change the SQL logic; it only
changes how the pieces are assembled:
# Single-cohort path:
result <- build_cohort_query_internal(cohort, opts)
sql <- result$full_sql
# Batch path (same builder, structured output):
result <- build_cohort_query_internal(cohort, opts)
# Uses result$codeset_union_parts, result$primary_events_sql,
# result$tail_sql to assemble into batch scriptThe structured output from the builder provides clean separation
points (codeset_union_parts,
primary_events_sql, tail_sql) rather than
relying on fragile regex extraction from generated SQL.
2. Domain Filtering Includes Source Concepts
Domain-filtered tables include both standard and source concept columns:
SELECT * INTO ##DRUG_EXPOSURE_FILTERED
FROM @cdm_database_schema.DRUG_EXPOSURE de
WHERE de.drug_concept_id IN (SELECT concept_id FROM #ALL_CONCEPTS)
OR de.drug_source_concept_id IN (SELECT concept_id FROM #ALL_CONCEPTS);This ensures rows that match only on
drug_source_concept_id (e.g., unmapped records with
drug_concept_id = 0) are not dropped in batch but retained,
matching the single-cohort behavior which queries the full CDM
table.
3. QualifiedLimit Preservation
When a cohort specifies QualifiedLimit: "First", the
single-cohort SQL includes WHERE QE.ordinal = 1 to select
only the first qualifying event per person. The Phase 1 rewrite must
preserve this filter. The assembler explicitly checks:
# In assemble_batch_script():
if (isTRUE(structured$has_qualified_limit) &&
!grepl("ordinal\\s*=\\s*1", ins2, perl = TRUE)) {
next # Skip Phase 1 for this cohort; run full script instead
}If the rewritten INSERT does not contain the ordinal = 1
filter, the cohort is routed through the full script
path rather than the Phase 1 + tail path, preserving correct
semantics.
4. Automated Equivalence Validation
The package includes validate_batch_equivalence() which
runs both paths on the same CDM and compares results row-by-row:
# Run batch path
cdm <- generateCohortSet2(cdm, cohortSet, name = "batch_test")
# Run each cohort independently (optimize=FALSE)
for (each cohort) {
atlas_json_to_sql_batch(single_cohort, optimize = FALSE)
}
# Compare per cohort_definition_id:
# (subject_id, cohort_start_date, cohort_end_date) must match exactlyThe test suite (test-batch-equivalence.R) verifies:
- Structured output completeness (full_sql, tail_sql, primary_events_sql, codeset_union_parts)
- Phase 1/Phase 2 generation for multi-cohort batches
- QualifiedLimit preservation in Phase 1 inserts
- Concept set exclusion handling (
isExcluded,LEFT JOIN ... IS NULL) - Mapped concept handling (
includeMapped,concept_relationshipjoin) - Row-level equivalence on a synthetic CDM (when CDMConnector is available)
Safety Mechanisms
Several additional safety checks prevent silent divergence:
-
can_vectorizetracking: A boolean per cohort that must be TRUE for the Phase 1 + tail path to be used -
EndStrategy detection: Cohorts with CustomEra or
DateOffset end strategies that reference
#strategy_endsskip Phase 1 and run the full script -
Unresolved parameter warnings:
apply_batch_rewrites()flags any@-parameters that survive rewriting (except known-safe ones like@vocabulary_database_schema) -
force_full_pathoption: Settingoptions(atlasCohortGenerator.force_full_path = TRUE)disables Phase 1 entirely and routes all cohorts through the full script path, useful for debugging
Performance Characteristics
The speedup from batch optimization depends on the workload. The main factors are: how many cohorts share vocabulary/domains, the size of the CDM, and result-table I/O cost.
By Workload Type
Independent Cohorts (Different Domains)
Cohorts that use entirely different CDM domains (e.g., one uses only
DRUG_EXPOSURE, another only MEASUREMENT) still
benefit from:
- Shared vocabulary expansion: 1 CONCEPT_ANCESTOR join instead of N
- Shared result-table I/O: 1 finalize instead of N
- No domain-scan savings: Each domain is only used by one cohort, so filtering happens once either way
Typical speedup: 1.5–3x for 5–20 cohorts. The savings come primarily from vocabulary and I/O consolidation.
Independent Cohorts (Overlapping Domains)
Cohorts that share CDM domains (e.g., multiple drug-exposure cohorts, or multiple condition-based cohorts) see the largest benefit:
-
Domain tables scanned once: A 500M-row
DRUG_EXPOSUREtable is filtered once into##DRUG_EXPOSURE_FILTERED, then all cohorts read from the smaller filtered table - Concept overlap: If cohorts share concepts (common in phenotype libraries), the filtered tables are even smaller
Typical speedup: 3–10x for 10–50 cohorts sharing 2–3 domains. The savings grow roughly linearly with N because each additional cohort avoids a full CDM scan.
Subset Cohorts (Same Primary Criteria, Different Inclusion Rules)
This is the best case. Subset cohorts (e.g., “all aspirin users” vs. “aspirin users with diabetes” vs. “aspirin users over age 65”) share:
-
Identical primary events: Phase 1 builds primary
events for each, but they read from the same
##DRUG_EXPOSURE_FILTERED - Same vocabulary: Often the exact same concept sets
-
Phase 2 is cheap: Only inclusion rules differ;
slicing from
#QUALIFIED_EVENTS_ALLis fast
Typical speedup: 5–15x for 5–20 subset cohorts. The primary-events build (the most expensive phase) operates on pre-filtered data, and Phase 2 per-cohort blocks do minimal additional work.
Large Phenotype Libraries (100+ Cohorts)
For large-scale studies generating 100+ cohorts:
- All vocabulary expansion is done once (can save minutes on large vocabularies)
- Domain tables are filtered once (can save tens of minutes for billion-row CDMs)
- Result-table I/O is a single bulk operation
Typical speedup: 5–20x depending on CDM size and cohort diversity.
Summary Table
| Workload | N | Shared Domains | Speedup Range |
|---|---|---|---|
| Independent, different domains | 5–20 | Low | 1.5–3x |
| Independent, overlapping domains | 10–50 | High | 3–10x |
| Subset cohorts | 5–20 | Very high | 5–15x |
| Large phenotype library | 100+ | Mixed | 5–20x |
The optimizer adds negligible overhead for small batches (< 5 cohorts) and never produces slower results than the one-at-a-time approach.
Code Example
Single Cohort (No Optimization)
library(atlasCohortGenerator)
# Generate SQL for one cohort
sql <- atlas_json_to_sql(
json = "path/to/aspirin_users.json",
cohort_id = 1,
cdm_schema = "cdm",
target_schema = "results",
target_dialect = "sql server"
)Batch (Optimized)
# Generate optimized batch SQL for multiple cohorts
batch_sql <- atlas_json_to_sql_batch(
json_inputs = list(
"path/to/aspirin_users.json",
"path/to/metformin_users.json",
"path/to/aspirin_diabetics.json"
),
cdm_schema = "@cdm_database_schema",
results_schema = "@results_schema",
target_dialect = "sql server",
optimize = TRUE # default
)
# Or with a cohort set data frame (e.g. from CDMConnector::readCohortSet)
cohort_set <- data.frame(
cohort_definition_id = c(1, 2, 3),
cohort = c("aspirin.json", "metformin.json", "aspirin_diabetics.json")
)
batch_sql <- atlas_json_to_sql_batch(
json_inputs = cohort_set,
optimize = TRUE
)With CDMConnector
library(CDMConnector)
library(atlasCohortGenerator)
cdm <- cdm_from_con(con, cdm_schema = "cdm", write_schema = "results")
cohort_set <- readCohortSet("path/to/cohort/jsons/")
# generateCohortSet2 uses the batch optimizer automatically
cdm <- generateCohortSet2(cdm, cohort_set, name = "my_cohorts")Validating Equivalence
# Compare batch vs single-cohort results on your CDM
ok <- validate_batch_equivalence(cdm, cohort_set, name = "equiv_test")
# Prints per-cohort match status; returns TRUE if all matchDebugging
# Dump the final batch SQL for inspection
options(atlasCohortGenerator.debug_sql_file = "batch_debug.sql")
cdm <- generateCohortSet2(cdm, cohort_set, name = "debug_run")
# Force all cohorts through the full (non-optimized) script path
options(atlasCohortGenerator.force_full_path = TRUE)
cdm <- generateCohortSet2(cdm, cohort_set, name = "full_path_run")Appendix: How the DAG Is Enforced
The batch optimizer does not use an explicit graph data structure. Instead, the DAG is enforced by SQL execution order: the assembled script is a linear sequence of SQL statements where each phase only references tables created in earlier phases. There are no back-edges (later phases never write to tables read by earlier phases) and no cross-cohort dependencies (no cohort’s Phase 2 block reads from another cohort’s Phase 2 output).
This linear ordering is a valid topological sort of the implicit DAG:
#CODESETS_GLOBAL
|
v
#ALL_CONCEPTS
|
+----> ##DRUG_EXPOSURE_FILTERED
+----> ##CONDITION_OCCURRENCE_FILTERED
+----> ##PROCEDURE_OCCURRENCE_FILTERED
+----> ... (other domains)
+----> ##ATLAS_OBSERVATION_PERIOD
|
v
#QUALIFIED_EVENTS_ALL (Phase 1: per-cohort inserts, shared table)
|
v
Per-cohort Phase 2 blocks (independent of each other)
|
v
Staging tables (#COHORT_STAGE, ...)
|
v
Finalize (results tables)Because Phase 2 blocks are independent, they could in principle be parallelized. The current implementation executes them sequentially within a single SQL batch, but the DAG structure means no ordering constraint exists between different cohorts’ Phase 2 blocks.